

Les surdités neurosensorielles de l’enfant sont en grande majorité d’origine cochléaire. Nous aborderons en fin de chapitre les neuropathies auditives. Jusqu’à ces dernières années, on estimait que :
- Un tiers des surdités de l’enfant était de cause environnementale,
- Un tiers seulement avait une origine génétique,
- Un tiers était de cause indéterminée.
On sait maintenant que la majorité des cas sporadiques de surdité congénitale sont des formes génétiques récessives de surdité. Les causes extrinsèques semblant moins fréquentes dans les études les plus récentes, on peut estimer que près de ¾ des surdités congénitales sont donc d’origine génétique.
La recherche étiologique dans les surdités de perception est fondée avant tout sur un interrogatoire et un examen clinique très détaillés. Sauf cas particulier, il est souhaitable de dissocier l’enquête étiologique de l’annonce de la surdité et d’attendre quelques mois que la prise en charge audio prothétique, orthophonique et éducative soit mise en place.
Les causes extrinsèques ou environnementales
Dans la population occidentale, 27% à 43,5% des surdités de l’enfant sont dues à des facteurs environnementaux (surdités acquises pré, péri ou post natales).
- Les causes prénatales
Les causes prénatales représentent en moyenne 11% de l’ensemble des surdités de l’enfant. La rubéole congénitale est le plus souvent en cause dans ces séries, mais en forte baisse depuis plusieurs années.
L’infection pré natale à cytomégalovirus (CMV) est probablement largement sous estimée. On sait en effet que non seulement la primo infection maternelle mais aussi les réinfections à CMV peuvent être cause de surdité, surtout unilatérale et dans un contexte infectieux beaucoup moins évident à diagnostiquer chez le nouveau-né en cas de ré-infections.
Les études récentes systématiques de virurie chez le nouveau-né montrent que cette exposition prénatale au CMV est fréquente : on etime que chaque année en France, 2000 infections congénitales sont dues à une primo-infection à CMV et 3300 à une réinfection maternelle. Parmi ces 5300 cas, 200 nouveaux-nés seulement seront symptomatiques à la naissance, avec une surdité dans plus de la moitié des cas. Mais environ 7% des nouveaux-nés asymptomatiques auront une surdité séquellaire (parfois d’apparition secondaire ou évolutive).
Quelle est la valeur rétrospective d’une sérologie CMV ou d’une recherche de virurie à CMV ? En cas de surdité diagnostiquée les premiers mois de vie, ces examens sont utiles, car une virurie peut persister plusieurs mois après la naissance et faire le diagnostic d’infection pré natale à CMV. Mais chez le grand enfant, une sérologie CMV positive a peu de valeur diagnostique rétrospective, car le pourcentage de séropositivité au CMV augmente rapidement avec l’âge.
Les autres infections prénatales, comme la toxoplasmose, sont plus rares de même que l’exposition à des médicaments ototoxiques (aminoglucides, furosémide) pendant la grossesse.
- Les surdités de cause périnatale
Les surdités de cause périnatale représentent environ 14% des surdités de l’enfant. Elles sont dues à la combinaison de plusieurs facteurs, qui font partie de la liste des facteurs de risque de surdité.
-> Poids de naissance < 2000 grammes et/ou âge gestationnel < 34 semaines ;
-> Asphyxie néonatale sévère, avec APGAR < 4 à 5 minutes ;
-> Pathologie respiratoire néonatale sévère (FIO2 élevé, ventilation mécanique de plus de 12 heures) ;
-> Traitement ototoxique (aminosides, furosémide, etc.) ;
-> Hyperbilirubinémie ayant nécessité une exsanguino-transfusion.
Ces facteurs de risque doivent faire pratiquer impérativement un bilan auditif. Prouver l’implication d’un de ces facteurs de risque est très souvent impossible. Il faudra toujours envisager la possibilité d’une surdité génétique congénitale et proposer une consultation de conseil génétique.
- Les surdités post natales
Les surdités post natales représentent 11% des surdités. Elles sont acquises après la naissance et durant l’enfance. Elles sont dues en majorité à des méningites bactériennes (à haemophilus influenzae et streptococcus pneumoniae essentiellement) et à l’administration de médicaments ototoxiques (streptomycine, gentamycine, furosémide). On retrouve également d’autres causes plus rares comme les oreillons, les labyrinthites infectieuses, les otites chroniques, les fractures du rocher, les traumatismes sonores ou pressionnels et les exceptionnelles causes tumorales.
Dans certains cas, le caractère acquis de la surdité peut être affirmé : c’est le cas de certaines surdités apparues durant l’enfance, post méningitique, toxique ou traumatique. De même pour la toxoplasmose, la surveillance sérologique des grossesses permet souvent un diagnostic précis. Cependant, pour la majorité des causes extrinsèques, il est difficile d’être certain de l’étiologie et le bilan étiologique devra être effectué comme pour une surdité de cause inconnue. C’est le cas de l’anoxie néonatale, la prématurité, l’ictère nucléaire, la prise de toxiques en période pré ou périnatale.

Les surdités de perception d’origine génétique
Les surdités génétiques sont, dans la grande majorité des cas, des maladies mono géniques : l’atteinte d’un seul gène est en cause dans chaque forme de surdité et la déficience auditive est due à une atteinte cochléaire dans la très grande majorité des cas. De nombreux gènes responsables de surdité sont actuellement localisés sur les chromosomes humains : plus de 70 sont responsables de formes non syndromiques (la surdité étant la seule manifestation de l’atteinte génique) et plus de 100 sont impliqués dans des surdités syndromiques. Plusieurs de ces gènes sont impliqués dans une forme syndromique et non syndromique.
-
Modes de transmission
Les surdités génétiques peuvent se transmettre selon plusieurs modes. Pour les gènes de surdité autosomiques (situés sur les chromosomes non sexuels), selon la fonction et le type de mutation du gène, on peut observer :
• Une transmission récessive : les deux allèles du gène doivent être mutés pour que le sujet soit sourd ;
• Une transmission dominante : les sujets ayant un seul allèle muté (hétérozygotes) sont sourds, par exemple parce que la mutation génétique de l’allèle atteinte produit une protéine anormale, qui modifie la fonction de la protéine normale codée par l’allèle sain.

Les modes de transmission sont :
• Autosomique récessif (AR) : dans ce mode de transmission, les deux parents sont normoentendants, porteurs d’une copie anormale (allèle) du gène en cause et statistiquement un quart des enfant (garçon ou fille) sont sourds, porteurs de mutations sur les deux allèles du gène. Le mode de transmission autosomique récessif est favorisé par la consanguinité. On estime qu’environ ¾ des surdités non syndromiques se transmettent sur le mode AR et ce mode est le deuxième en fréquence dans les surdités syndromiques.
• Autosomique dominant (AD) : dans les familles atteintes de surdité autosomique dominante, l’un des parents est sourd et porte sur un seul allèle du gène la mutation pathogène, mutation qu’il va transmettre à la moitié de ses enfants qui seront alors sourds. L’expressivité est très souvent variable dans ce mode de transmission, plusieurs sujets atteints dans la famille pouvant présenter des surdités de sévérité très différente et surtout lorsque la surdité est syndromique, les signes associés au syndrome peuvent être absents ou discrets chez certains sourds de la famille, chez qui la surdité paraît alors isolée.
• Lié à l’X : le gène en cause est situé sur le chromosome X. Chez les garçons, qui n’ont qu’un X, la maladie s’exprime et ils sont donc atteints de surdité. Ils transmettront l’X porteur de la mutation génétique à leurs filles. Chez les femmes, l’X porteur de la mutation génétique est en général « compensé » par le deuxième X normal (surdité dite récessive liée à l’X). Rarement, comme dans le syndrome d’Alport, la surdité est dite dominante lié à l’X et les femmes ne sont alors pas seulement transmettrices mais aussi atteintes de la surdité, de façon moins sévère que les garçons.
• Le mode de transmission mitochondrial : le génome mitochondrial est un fragment d’ADN situé hors du noyau de la cellule, dans la mitochondrie. Il est transmis par la mère. Lorsqu’un gène de surdité est situé sur l’ADN mitochondrial, l’arbre généalogique est caractéristique car hommes et femmes peuvent être sourds, mais seules les femmes pourront transmettre la surdité à leurs enfants, qui sont, en théorie, tous sourds dans la fratrie.
-
Les surdités syndromiques
Les surdités syndromiques ne rendent compte que d’une faible proportion des surdités de l’enfant (10 à 15% environ). Plusieurs centaines de syndromes avec surdité ont été décrits et plus d’une centaine de gène sont identifiés à ce jour. Il est cependant important de connaître et rechercher les principaux syndromes car la prise en charge et le bilan étiologique seront différents d’une surdité non syndromique. En raison du très grand nombre de syndromes rares avec surdité, toute pathologie malformative chez l’enfant doit faire pratiquer un bilan auditif systématique (otoémissions acoustiques les premiers mois de vie ou tests subjectifs). De plus, pour les surdités syndromiques comprenant une atteinte malformative cranio-faciale, la surdité est très souvent majorée par une otite chronique et la surveillance otologique régulière s’impose. Certains syndromes sont particulièrement importants à connaître en raison de leur fréquence et/ou de leur gravité potentielle :
Trois syndromes autosomiques récessifs doivent être systématiquement recherchés car ils se présentent initialement comme une surdité isolée : les syndromes de Pendred et de Usher, tous deux fréquents et le rare syndrome de Jervell et Lange-Nielsen. Tous ces syndromes ont la particularité de se présenter longtemps comme une surdité isolée et seul un bilan systématique peut permettre de les détecter précocement.
- Le syndrome de Pendred
Ce syndrome a été décrit il y a plus de cent ans. Il est reconnu par l’association d’une surdité d’origine cochléaire le plus souvent évolutive, pré linguale ou post linguale précoce, à un trouble de l’organification de l’iode qui se manifeste par un goitre thyroïdien. Il se transmet sur un mode récessif autosomique. Le gène en cause, PDS (maintenant appelé SLC26A4) est impliqué à la fois dans le syndrome de Pendred et une forme de surdité qui reste isolée DFNB4. La frontière entre le syndrome de Pendred et la forme de surdité isolée DFNB4 est parfois difficile à définir, puisque au sein de familles atteintes de Pendred, un ou plusieurs individus peuvent ne pas développer l’atteinte thyroïdienne.
La prévalence du syndrome de Pendred est estimée à 7-10 cas sur 100 000 naissances. La surdité a la particularité, lorsqu’elle n’est pas profonde d’emblée, d’évoluer par paliers d’aggravation brutale suivis d’une récupération en général partielle. Ces fluctuations sont extrêmement handicapantes et angoissantes pour l’enfant.
L’âge d’apparition du goitre thyroïdien est variable, le plus souvent au cours de la deuxième décennie (70% des patients n’ont pas de goitre à 10 ans). Le syndrome de Pendred se présente donc comme une surdité isolée pendant de nombreuses années. Le goitre s’associe à une hypothyroïdie dans 50% des cas.
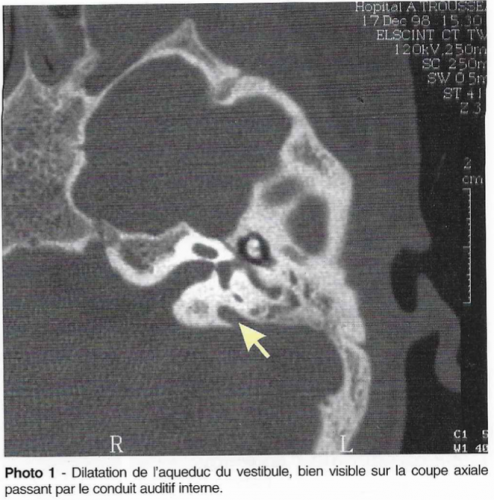
Le scanner des rochers met en évidence des anomalies morphologiques de l’oreille interne (dilatation de l’aqueduc du vestibule), cochlée incomplète de type « Mondini » de façon quasi constante, mais ces anomalies peuvent être unilatérales. Les anomalies d’organification de l’iode peuvent être mises en évidence par la scintigraphie thyroïdienne avec test au perchlorate : l’incorporation des iodures à la molécule de thyroglobuline se fait de façon anormale et l’administration de perchlorate, anion anorganique, induit un relargage des iodures non organifiées. La mesure de la quantité d’iode radioactive avant et après administration de perchlorate montre une diminution supérieure à 10%. Ce test peut permettre de détecter un syndrome de Pendred avant l’apparition du goitre chez les patients avec malformation de l’oreille interne. Cependant, ce test n’est ni sensible (normal chez certains sujets dans des formes familiales) ni spécifique (positif notamment dans la thyroïdite de Hashimoto et chez les patients avec mutations des gènes de la thyroïde peroxydase ou de la thyroglobuline). Il a, de plus, l’inconvénient d’être irradiant.
Le gène PDS code pour la pendrine formée de 780 acides aminés. A ce jour, cinquante mutations dans le gène PDS ont été identifiées. Quatre d’entre elles sont particulièrement fréquentes, présentes sur 74% des chromosomes mutés.
- Le syndrome de Usher
Ce syndrome associe à la surdité une rétinite pigmentaire. Il existe de multiples formes de syndromes de Usher, mais les ¾ sont des Usher de type I avec surdité congénitale profonde, aréflexie vestibulaire bilatérale responsable d’un retard à la marche (marche après 18 mois) et rétinite qui se développe pendant l’enfance. Les premiers signes visuels sont des troubles de la vision dans la pénombre, souvent vers 10 ans, mais le fond d’œil systématique peut faire le diagnostic bien avant cet âge, dès 3-4 ans. L’examen le plus précoce est l’électrorétinogramme, pathologique avant les premiers signes au fond d’œil, mais examen invasif et désagréable. Le syndrome de Usher de type I est une indication d’implant cochléaire précoce pour obtenir une compréhension du langage sans lecture labiale chez ces enfants qui vont devenir sourds aveugles à l’âge adulte. Faire le diagnostic par le fond d’œil à 4 ans est donc déjà tard. En pratique, l’examen ophtalmologique avec fond d’œil doit être systématique et répété chez l’enfant et l’adulte sourd et toute surdité profonde congénitale avec retard à la marche sans étiologie évidente doit faire pratiquer un électrorétinogramme, même si le fond d’œil est normal.
Dans le syndrome de Usher type II, la surdité est de moyenne-sévère non progressive, prédominante sur les fréquences aigues, la rétinite un peu plus tardive et les signes vestibulaires absents. Dans le Usher de type III, la surdité est progressive, les signes vestibulaires et l’âge de début de la rétinite sont variables.
A ce jour, 11 gènes ont été localisés et 6 de ces gènes sont identifiés. 4 pour le syndrome de Usher de type I, 1 pour le type II et 1 pour le type III. MYO7A et CDH23 représentent respectivement 30% et 29% des cas de Usher de type I et USH2A 40% des type II. Le diagnostic moléculaire n’est pas fait en routine et le diagnostic est essentiellement clinique.
- Le syndrome de Jervell et Lange-Nielsen
Ce syndrome est rare mais de diagnostic facile par un ECG systématique en cas de surdité sévère ou profonde congénitale. L’ECG montre un allongement de l’espace QT qui traduit un trouble de conduction cardiaque, source de malaise ou mort subite dont la prévention est possible par un traitement médical. 2 gènes sont identifiés.
Deux syndromes autosomiques dominants doivent être connus des ORL car fréquentes et probablement sous diagnostiqués : le syndrome de Waardenburg et le syndrome Branchio-Oto-Rénal.
- Le syndrome de Waardenburg
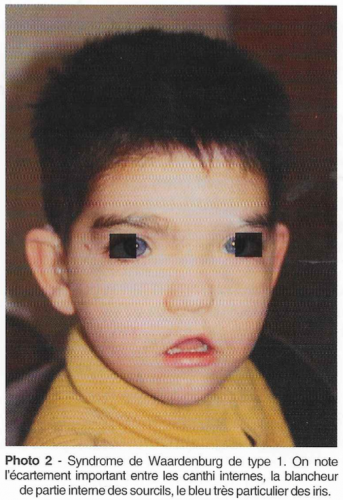
Il associe une surdité à des anomalies de pigmentation des cheveux (mèches blanches), des yeux (yeux très bleus, vairons, dépigmentation du fond d’œil), de la peau (tâches cutanées). Dans certaines formes (Waardenburg type I), il existe un écartement anormal entre les yeux, les canthi interne étant décalés vers l’extérieur avec diminution de longueur de la fente palpébrale (dystopie canthale). La surdité est très variable, uni ou bilatérale, légère à profonde, avec ou sans malformation de l’oreille interne. Le piège pour le diagnostic est que les particularités physiques qui peuvent exister dans la famille du sourd et être très évocatrices, ne sont pas spontanément décrites à l’interrogatoire, n’étant pas considérées comme des pathologies. Les mèches blanches sont de plus très souvent teintes. La question de l’existence d’yeux vairons ou de mèches blanches dans la famille doit donc être posée systématiquement.
4 types cliniques de syndrome de Waardenburg ont été décrits en fonction des signes associés :
- Le type I est associé à une dystopie canthale ;
- Le type II sans dystopie canthale ;
- Le type III (ou Klein-Waardenburg) est un type I associé à des malformations des extrémités ;
- Le type IV (ou Waardenburg-Shah), de transmission autosomique récessive, est un type II avec maladie de Hirschsprung.
6 gènes sont identifiés.
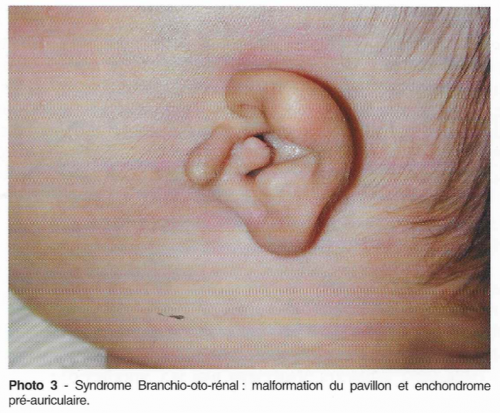
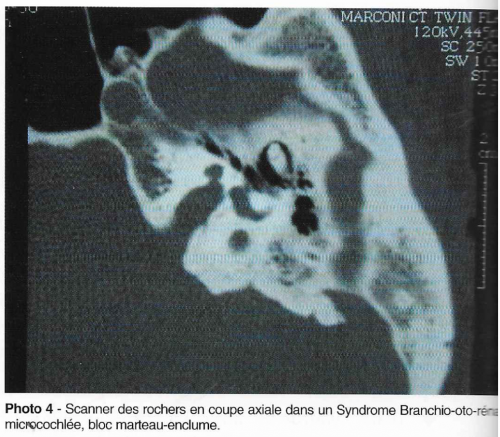
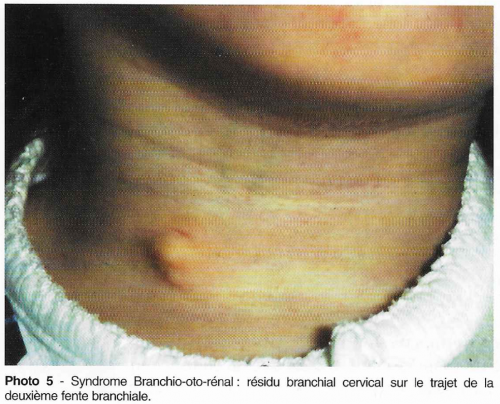
- Le syndrome Branchio-Oto-Rénal (BOR)
Ce syndrome associe une surdité, des fistules branchiales multiples et une malformation rénale. Les malformations rénales peuvent être majeures (agénésies ou hypoplasies majeures) et conduisent le plus souvent à une interruption de grossesse. Les malformations moins importantes seront diagnostiquées par une échographie rénale qui doit être demandée devant une surdité évocatrice du BOR : la surdité s’accompagne de malformations de l’oreille externe (oreilles mal ourlées, aplasies d’oreille, enchondromes, sténose des conduits auditifs), de l’oreille moyenne (il existe une composante transmissionnelle à l’audiogramme) et de l’oreille interne (diverses malformations cochléo vestibulaires). On retrouve en général des fistules préhélicéennes bilatérales et des fistules de la deuxième fente branchiale avec résidus cartilagineux associés évocateurs. En pratique, devant une surdité de perception ou mixte associée à une fistule branchiale ou à des malformations de l’oreille externe, il est souhaitable de faire une échographie rénale. 2 gènes ont été localisées et l’un d’eux est identifié : EYA1.
- Un syndrome lié à l’X, Le syndrome d’Alport
Il doit être recherché systématiquement pour toute surdité post linguale. La surdité est progressive (1ère décennie) associée à des épisodes d’hématurie (l’hématurie est d’abord microscopique pendant plusieurs années). La bandelette urinaire systématique chez l’enfant sourd permet un diagnostic et une prise en charge précoce de ce syndrome qui aboutit à l’insuffisance rénale à un âge variable, entre 30 et 50 ans. L’examen ophtalmologique retrouve un lenticone antérieur, évocateur de ce syndrome. L’Alport est un des rares syndromes dominants liés à l’X, ce qui veut dire que les femmes transmettrices ont une atteinte a minima autosomique récessive. Plusieurs gènes sont identifiés.
Les surdités non syndromiques
Il peut s’agir de cas sporadiques ou familiaux. Les formes autosomiques récessives sont les plus fréquentes et la surdité est en général congénitale. Dans les formes dominantes, la surdité est le plus souvent progressive ou d’apparition retardée, au cours de l’enfance ou à l’âge adulte. Dans ce domaine, les avancées en génétique moléculaire ont été très importantes depuis 1994 avec la localisation de plus de 70 gènes responsables chacun d’une forme de surdité isolée.
Nous n’aborderons ici que les formes les plus fréquentes.
-
Formes autosomiques récessives fréquentes (DFNB)
- La forme majoritaire de surdité de l’enfant DFNB1 (gène de la connexine 26)
On sait depuis 1997 que la forme de surdité autosomique récessive DFNB1, due à l’atteinte du gène de la connexine 26 (CX26 ou GJB2) est en cause dans la moitié des surdités récessives congénitales et 30% à 40% des cas sporadiques congénitaux en France. En effet, au vu de la petite taille des fratries en France, beaucoup de surdités dues à des mutations de CX26 se présentent comme des cas isolés.
Dans cette forme de surdité (appelée forme DFNB1), la déficience auditive est congénitale, peu ou pas progressive, de tous degrés mais le plus souvent profonde, avec des courbes audiométriques plates ou descendantes. La tomodensitométrie des rochers et les épreuves vestibulaires caloriques sont normales.
En France comme dans les pays occidentaux et méditerranéens, une mutation prédomine largement : 35delG. Les porteurs hétérozygotes (normo-entendants) de cette mutation sont très fréquents dans la population générale, entre 2,5% et 4% de la population en France, Espagne et Italie, 2% aux Etats-Unis. La prévalence des mutations de CX26 est comparable dans beaucoup de pays à celle du gène CFTR de la mucoviscidose. Un diagnostic moléculaire de routine est disponible dans de nombreux laboratoires en France. D’autres mutations majoritaires sont retrouvées dans certaines populations : 167delT dans la population juive ashkénaze, 235delC au Japon, en Chine et en Corée, R143W en Afrique noire.
La mise en évidence de mutations pathogènes sur les deux allèles de CX26 devant un cas sporadique de surdité congénitale permet d’affirmer le caractère génétique de la surdité, et de donner aux familles le risque de récurrence. Dans certaines formes familiales, le diagnostic moléculaire de mutations de CX26 peut permettre de préciser un mode de transmission peu clair.
- Les autres gènes de connexine
D’autres gènes de connexine peuvent être responsables d’une surdité isolée : GJA1, GJB6, GJB3 codant respectivement pour les connexines 43, 30 et 31. L’un de ces gènes, GJB6, est particulièrement important : très proche du gène de la connexine 26, une décision d’un seul allèle de ce gène est fréquemment retrouvée en association avec une mutation hétérozygote du gène de la connexine 26 chez des sujets sourds congénitaux.
- La surdité isolée DFNB4
Le gène du syndrome de Pendred, PDS, peut être responsable de la forme de surdité DFNB4, qui ressemble en tous points à celle du syndrome (prélinguale, progressive ou fluctuante, malformation de l’oreille interne) mais qui reste isolée, c’est à dire sans atteinte thyroïdienne. En 1998 et 1999, deux études ont montré l’implication de mutations de PDS dans la forme de surdité DFNB4, chez les sujets présentant une surdité congénitale profonde neurosensorielle sans goitre ni hypothyroïdie, mais avec une malformation de l’oreille interne au scanner des rochers (dilatation bilatérale de l’aqueduc du vestibule, qui contient le sac endolymphatique, avec pour critère un aqueduc large de plus de 2mm à sa partie moyenne). Le test au perchlorate était normal chez les sujets testés.
Campbell et al. (2001) ont montré que PDS était très fréquemment impliqué dans des formes familiales de surdité avec malformation de l’oreille interne : des mutations de ce gène sont mises en évidence chez 4/5 familles avec malformation de Mondini et 5/6 familles avec DAV.
La reconnaissance des surdités dues à PDS est importante pour le conseil génétique mais aussi pronostique car leur évolutivité prévisible peut aider l’orientation éducative et faire prendre des précautions vis à vis des microtraumatismes (certains sports, traumatisme pressionnels) chez ces enfants.
- DFNB9
Cette forme de surdité récessive est due à des mutations du gène de l’Otoferline, OTOF. L’otoferline serait impliquée dans le trafic vésiculaire pré synaptique des cellules ciliées internes. La surdité est sévère ou profonde, prélinguale et une mutation de ce gène, Q829X, semble particulièrement fréquente en Espagne. La particularité de cette forme de surdité est qu’elle peut se présenter comme une neuropathie auditive avec otoémissions acoustiques conservées.

-
Les surdités autosomiques dominantes, DFNA
Nous détaillerons quelques formes dont le gène est identifié et qui semblent assez fréquentes.
- DFNA2 est une forme de surdité progressive, qui atteint de façon prédominante les fréquences aigues initialement. L’âge de début est variable, entre un et trente ans, la surdité progresse d’environ 1dB par an et les acouphènes sont fréquents. Le gène en cause, KCNQ4, est exprimé dans les cellules ciliées externes et peut être internes et code pour un canal potassique voltage dépendant.
- DFNA9 est due à l’atteinte du gène COCH, exprimé en grande quantité dans la cochlée. Ce gène code pour une protéine de la matrice extra cellulaire, la cochline et les analyses histologiques des rochers montrent des dépôts acidophiles cochléaires très caractéristiques. La surdité commence sur les fréquences aigues à l’adolescence ou chez l’adulte et progresse rapidement pour atteindre toutes les fréquences. Certains patients ont des épisodes de vertiges, plénitude de l’oreille et acouphènes qui peuvent évoquer une maladie de Ménière, même si la forme de courbe est différente.
-
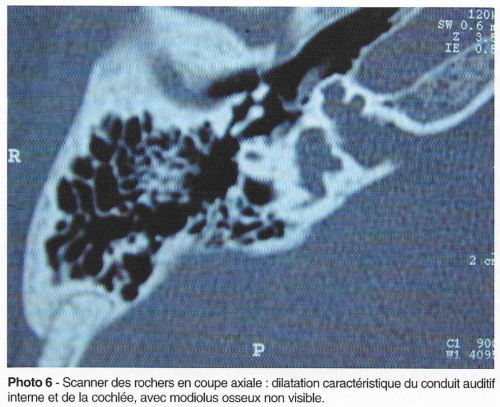
Les surdités liées à l’X, DFN
Beaucoup plus rares, seule une forme est génétiquement identifiée et particulièrement importante à connaître, surtout pour les otologistes : la surdité mixte liée à l’X avec geyser-labyrinthe (DFB3) due à l’atteinte du gène POU3F4. La surdité de perception s’accompagne d’une part transmissionnelle importante, avec Rinne de 50-60dB, qui a souvent, avant que ce syndrome soit connu, fait suspecter un blocage ossiculaire et pratiquer une exploration d’oreille : la platinectomie entraine un geyser massif et aboutit à une cophose. On sait maintenant que toute intervention chirurgicale pour surdité de transmission fixée chez l’enfant doit être précédée d’un scanner des rochers, qui montre dans ce syndrome une dilatation majeure du conduit auditif externe ainsi qu’une dilatation cochléovestibulaire.
-
Les surdités non syndromiques mitochondriales
Plusieurs mutations de l’ADN mitochondrial sont impliquées dans des surdités isolées : les mutations A1555G et 961delT de l’ARN ribosomal 12S et les mutations T7511C, A7445G et 7472insC de l’ARN de transfert de la sérine.
La surdité due à la mutation A1555G peut être de tout degré et apparaître à tout âge et a également été décrite chez des patients ayant développé une surdité après prise d’aminoglycosides. En effet, la forme d’ARN ribosomal 12S avec mutation A1555G lie les aminosides avec une affinité anormalement élevée, ce qui peut expliquer l’apparition de surdité pour les doses normales d’aminosides. A1555G est particulièrement fréquente en Espagne, dans les familles atteintes de surdité de transmission compatible avec une surdité mitochondriale, jusqu’à 25%.
Le bilan étiologique
Les éléments du bilan étiologique systématique des surdités neurosensorielles de l’enfant ne font pas actuellement l’objet d’un consensus dans la littérature.
Le principe du bilan étiologique est tout d’abord de rechercher, par interrogatoire, l’examen clinique et quelques examens complémentaires simples, une cause extrinsèque de surdité ou une pathologie associée qui pourrait orienter vers une surdité syndromique génétique.
Après la période initiale de diagnostic et de prise en charge de la surdité, le rôle de l’ORL est fondamental pour initier la recherche étiologique. Cette démarche permet de mieux informer les familles en collaboration avec un généticien et de prendre en charge les éventuelles pathologies associées.
Il paraît raisonnable de limiter les examens à visée étiologique à ceux résumés dans le tableau suivant. On peut aussi discuter, chez le nourrisson, la pratique d’une sérologie du cytomégalovirus, qui n’aura de valeur que négative pour éliminer cette étiologie. Positive, la sérologie ne pourra pas prouver que l’infection est bien prénatale, sauf si une virurie à CMV est mise en évidence (encore détectable les premiers mois de la vie).
Au terme du bilan, si une forme syndromique a été découverte, il faut organiser la prise en charge et le suivi et en cas de forme non syndromique, on peut s’orienter vers un diagnostic moléculaire avec recherche de mutations du gène de la connexion 26. En pratique, une consultation de conseil génétique est souhaitable dans tous les cas si le patient sourd et/ou sa famille sont demandeurs et beaucoup de généticiens en France sont maintenant formés à ce domaine particulier qu’est la génétique des surdités.

Les neuropathies auditives
Starr (1998) a, le premier, utilisé le terme de neuropathie auditive pour désigner chez l’adulte des surdités, souvent bilatérales, associées à une altération des PEA contrastant avec la présence d’otoémissions acoustiques (OEA) normales. Ces formes sont maintenant mieux connues chez l’adulte et l’enfant et justifient des pratiquer des otoémissions pour les individualiser, au cours de la prise en charge d’une surdité de perception.
La fréquence de ces surdités est très élevée (1%) chez les nouveaux-nés hospitalisés en Soins Intensifs, justifiant un dépistage de surdité par les potentiels évoqués plutôt que par OEA dans cette population. La prévalence ne serait que de 1/500 000 chez les nouveaux-nés non hospitalisés en Soins intensifs. Cependant, Madden (2002) a rapporté une prévalence plus élevée, 22 neuropathies sur 428 enfants sourds (0,5%), dans un centre hospitalier de référence aux USA (avec peut être un biais de recrutement). Parmi ces 22 cas pédiatriques, 15 (68%) avaient eu des pathologies néonatales intriquées (hyper bilirubinémie, prématurité, médicaments ototoxiques, ventilation assistée). 6 cas étaient 3 fratries de deux enfants sourds évoquant une cause génétique autosomique récessive. La surveillance du niveau auditif doit être rapprochée car des améliorations spontanées du seuil auditif peuvent survenir dans la moitié des cas, surtout en cas d’antécédents d’hyper bilirubinémie. Ces surdités peuvent bénéficier d’un appareillage conventionnel et Madden rapporte de bons résultats de l’implant cochléaire chez 2 enfants avec forme familiale.
—
Sources : les monographies du CCA Groupe, Surdité de l’enfant n°34, édition 2003
Erea-Noel Garabédian, Françoise denoyelle, René Dauman, Jean-Michel Triglia, Eric Tuy, Natalie Loundon, Patrick Bouaziz, Jean de Lorenzi








