

Les surdités par malformations congénitales
Les malformations congénitales de l’oreille sont essentiellement représentées par les aplasies majeures et mineures. L’ensemble des malformations congénitales de l’oreille visibles à la naissance représente 1 cas sur 2000 à 5000 naissances.
L’aplasie d’oreille est une malformation congénitale de l’oreille externe et/ou moyenne. On distingue :
- L’aplasie majeure, où la malformation congénitale du conduit auditif externe et/ou du pavillon est majeure et le plus souvent associée à une malformation de l’oreille moyenne ;
- L’aplasie mineure, où l’élément dominant est une malformation de l’oreille moyenne, parfois associée à une anomie mineure de l’oreille externe.
L’association d’une surdité de perception à une aplasie d’oreille est retrouvée dans 5% à 10% des cas, surtout dans un contexte de syndrome (Goldenhar, CHARGE, Branchio-oto-rénal, etc.)
L’étiopathogénie de ces aplasies reste mal connue. L’origine génétique est certaine dans certains syndromes connus et suspectée dans les formes isolées (10% à 14% d’anomalie de l’oreille externe retrouvées à l’enquête familiale). Certains agents tératogènes tels que le Thalidomide, la vitamine A et ses dérivés, peuvent interférer avec l’organogenèse de l’oreille moyenne et externe au cours du premier trimestre de grossesse. Les causes infectieuses sont très rares actuellement.
> L’aplasie majeure
Les aplasies majeures se rencontrent dans 1 cas sur 10 000 à 20 000 naissances. Les formes bilatérales représentent 20% à 30% des cas. Une aplasie mineure controlatérale est associée dans 3% à 5% des cas d’aplasie majeure unilatérale.
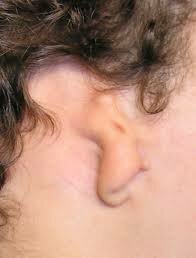
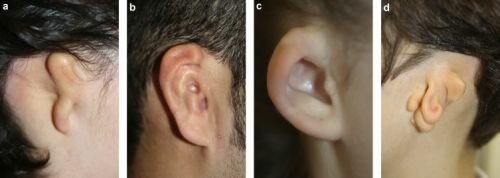
Les aplasies majeures sont des anomalies majeures du pavillon de l’oreille, en général associées à une malformation du conduit auditif externe et de l’oreille moyenne. Dans la forme la plus fréquente, le pavillon de l’oreille est réduit à un bourrelet chondrocutané verticalité et le conduit auditif est absent ou très malformatif. L’oreille en cornet est également assez fréquente, avec un conduit auditif le plus souvent présent et une microtie parfois corrigible par une otoplastie locale. Plus rares sont les microties importantes avec principaux reliefs identifiables, qui nécessitent une reconstruction avec cartilage costal, comme les formes classiques d’aplasie et les anoties, absence complète de pavillon et de conduit auditif, souvent associée à une implantation basse des cheveux qui gène la reconstruction.
Le premier examen, en période néonatale, précise l’aspect du pavillon, l’existence d’un conduit auditif externe, d’un tympan, de fistules cutanées. il faut rechercher une atteinte controlatérale, une malformation associée. Outre l’audiométrie, le bilan systématique comportera un examen ophtalmologique, une radiographie du rachis cervical, une échographie cardiaque et une échographie rénale.
La prise en charge initiale a pour but :
- D’apprécier le niveau auditif global, qui conditionnera la nécessité d’un appareillage auditif,
- De rechercher les malformations associées et une étiologie.
Le niveau auditif
Une aplasie majeure avec absence de conduit auditif externe entraine une surdité de transmission de 60 à 70dB. Dans les formes unilatérales, il est impératif d’éliminer une aplasie mineure contrôlable (otoémissions acoustiques ou potentiels évoqués auditifs). Pour les formes bilatérales, il est nécessaire de rechercher une surdité de perception associée par malformation de l’oreille interne. Dans tous les cas, on prévoira ultérieurement une audiométrie en champ libre, puis, dès que possible, oreilles séparées.
La recherche de malformations associes et le diagnostic étiologique
Les malformations faciales ou syndromes les plus fréquents sont :
- Un syndrome otomandibulaire (environ 20% des cas mais l’hypoplasie discrète de l’hémimandibule est presque constante),
- Une paralysie ou parésie faciale homolatérale,
- Une paralysie ou parésie de voile homolatérale,
- Un syndrome de Franceschetti, 3% des cas, avec notamment hypoplasie mandibulo-malaire bilatérale,
- Un syndrome de Goldenhar (oculo-auriculaire-vertébral),
- Un syndrome Branchio-Oto-Rénal.
Au terme du bilan néo-natal, l’appareillage auditif sera débuté dès les premiers mois si nécessaire. Il peut consister en un vibrateur mastoïdien sur serre-tête en cas d’aplasie bilatérale, ou en une conduction aérienne s’il existe un conduit auditif externe (au mieux par contour d’oreille). Dans certains cas, après reconstruction du pavillon, le vibrateur peut être porté sur des branches de lunettes. La BAHA (Bone Anchored Hearing Aid) est envisageable dès l’âge de 4 à 6 ans en relais du vibrateur ou si les appareillages auditifs conventionnels sont mal tolérés ou apportent un gain insuffisant.
L’orthophonie est indispensable dans les atteintes bilatérales dès le plus jeune âge, tout d’abord pour assurer la guidance parentale puis pour la réeducation orthophonique proprement dite.
La chirurgie fonctionnelle a pour but, dans les aplasies avec absence de conduit auditif externe, d’améliorer la surdité de transmission en recréant un conduit auditif externe au travers de la mastoïde, de recréer un tympan puis assurer la conduction du son jusqu’à la fenêtre ovale soit par la chaîne ossiculaire existante, soit par une prothèse. Cette chirurgie n’est pas toujours possible (procidence méningée, caisse non pneumatisée, nerf facial de trajet aberrant). Les résultats sont inconstants avec un seuil auditif post-opératoire jugé satisfaisant (de 0 à 25dB) dans 25% à 40% des cas. Les complications ne sont pas négligeables (paralysie faciale, otorrhée, sténose du néoconduit) et amènent à réserver cette chirurgie aux aplasies majeures bilatérales ou en cas de cholestéatome du conduit (survenant sur un conduit auditif malformé). Cette chirurgie fonctionnelle peut être envisagée dès 5 – 6 ans mais, pour la majorité des équipes, elle n’est plus indiquée dans les formes unilatérales en raison du mauvais rapport bénéficie-risque. Lorsqu’un conduit auditif est sténosé mais présent, les chances de succès sont meilleures et les risques de complication bien moindres : la décision chirurgicale pourra être prise après consentement éclairé de la famille et, si possible, de l’enfant.
La chirurgie de reconstruction du pavillon
Pour les lésions de type enchondromes, l’exérèse peut être pratiquée dès les premiers mois de vie, par simple ligature ou exérèse chirurgicale réglée. Pour la reconstruction du pavillon de l’oreille, la technique de Nagata en deux temps est actuellement utilisée. Elle donne de bons résultats esthétiques dans les formes les plus fréquentes, avec implantation des cheveux subnormale et absence d’antécédents chirurgicaux dans la zone de reconstruction. Le premier temps chirurgical est réalisé vers l’âge de 8 ans, avec un délai de quelques mois pour le deuxième temps.
> L’aplasie mineure
Définition
Il s’agit d’une malformation de l’oreille moyenne isolée ou associée à des malformations mineures de l’oreille externe (pavillon de l’oreille ou méat acoustique externe) ou encore à une malformation cervico-faciale ou générale.
Elle entraine typiquement une surdité de transmission ou une surdité mixte générale, avec courbe horizontale, non évolutive. Il n’y a pas d’acouphènes ni de vertige, ni de passé d’otite chronique. Il existe parfois des antécédents familiaux de surdité. Toutefois, le diagnostic n’est pas toujours évident :
- On peut retrouver une aplasie mineure associée à un tableau d’otite chronique et, ce, notamment dans de nombreux syndromes avec malformations cranio-faciale ;
- Il est parois difficile de retrouver à l’anamnèse, le caractère congénital et fixé de la surdité. En effet, les aplasies mineures isolées unilatérales peuvent être découvertes plus tard, vers l’âge de 5-6 ans.
Classification
Les différents types d’aplasie mineure sont très variables, touchant ou non la membrane tympanique ou secondaire à une malformation ou à un trouble de mobilité d’un ou de plusieurs osselets ou des fenêtres.
De nombreuses classifications ont été décrites, ce qui rend compte de la très grande diversité des anomalies rencontrées dans cette pathologie et de la difficulté d’établir des résultats prédictifs en fonction du type d’anomalie.
On retiendra 3 classifications qui ont été décrites après l’exploration chirurgicale de grandes séries de malformations mineures :
- La classification de Charachon, décrite en 1989 puis modifiée en 1994. Elle tient compte de l’origine embryologique de l’anomalie et de la possibilité d’une chirurgie fonctionnelle.

- La classification de Teunissen en 1991. Intéressante car réalisée à partir d’une série importante de cas et basée sur l’existence ou non d’un blocage platinaire, ce qui est essentiel dans la décision de la technique opératoire.

- La classification de Morisseau-Durand en 1994. C’est une variante de la classification de Charachon. Elle tient compte des anomalies platinaires pour chaque classe.

Circonstances de découvertes
Contrairement aux aplasies majeures, les aplasies mineures sont en général découvertes plus tardivement, en fonction de leur uni ou bilatéralité, de leur caractère isolé ou appartenant à un syndrome polymalformatif. Les formes bilatérales sont généralement diagnostiquées vers 2-3 ans, devant un retard de langage.
Une aplasie mineure doit systématiquement être recherchée devant :
- Une malformation mineure du pavillon (légèrement décollé, implanté plus bas que normalement et de plus petite taille, helix mal ourlé, lobule déformé) ou un enchondrome préauriculaire ;
- Une étroitesse ou anomalie d’orientation (en haut et arrière) du méat acoustique externe (dans 44% des cas des aplasies mineures) ;
- Un aspect réduit de la surface du tympan ou toute anomalie de sa mobilité ;
- Une aplasie majeure controlatérale ;
- Une fistule préhélicéenne ;
- Une fistule branchiale ;
- Une malformation cranio-faciale notamment : syndrome de Franceschetti, micrognathie même isolée, fente labiale ou vélopalatine, paralysie faciale congénitale, craniosténose, syndrome cervicoacoustique de Wildervank, maladie de Lobstein.
L’audiométrie et l’impédancemétrie
Les séries de Morisseau et Portmann décrivent un Rinne moyen de 42dB à 48dB. Pour Portmann, la CO est :
- Normale dans 45% des cas,
- Anormale dans les fréquences aigües dans 53% des cas, avec parfois une encoche sur les 2000Hz,
- Et l’écart entre fréquences conversationnelles (500 à 2000Hz) et le 4000Hz dépasse 20dB dans 18% des cas.
La courbe aérienne est horizontale sans encoche de Carhart, la surdité est non évolutive, le plus souvent unilatérale.
Le tympanogramme peut être normal ou mettre en évidence une hauteur de pic diminuée ou au contraire infinie (rupture de chaine). Le réflexe stapédien est aboli du côté atteint en ipsi et controlatéral dans la majorité des cas.
Imagerie
Le scanner des rochers en coupe millimétriques axiales et coronales sur un appareil de troisième génération va permettre de mettre en évidence les anomalies des corps des osselets, les blocages atticaux, la dysplasie des fenêtres, les anomalies de trajet des deuxième et troisième portion du nerf facial. Par contre, il est toujours difficile de détecter les anomalies de la branche descendante de l’enclume ou de la superstructure de l’étrier, sauf malformation importante.
Le scanner est l’examen clé du diagnostic et permet d’évaluer l’anatomie de l’oreille en vue d’une intervention chirurgicale, de rechercher une anomalie de l’oreille interne (surtout en cas de surdité mixte) ou du trajet du nerf facial qui interdirait toute ouverture platinaire ou toute reconstruction.
Traitement
Tous les auteurs s’accordent pour dire que les résultats fonctionnels des chirurgies de l’aplasie mineure sont loin d’être constants (40% à 48% des résultats bons et moyens pour Morisseau-Durand et Charachon). Les contre-indications opératoires, outre certains risques anesthésiques, sont :
- Certaines anomalies du trajet nerf facial empêchant tout reconstruction,
- Les anomalies de l’oreille interne ou du conduit auditif interne. Ces malformations entrainent un risque de geyser de périlymphe lors de l’ouverture platinaire avec ses conséquences sur l’audition (cophose immédiate ou retardée) et un risque de méningite.
Selon Morisseau-Durand, il est important de suspecter une anomalie de l’oreille interne devant toute surdité mixte à courbe descendante et, a fortiori, devant une altération de plus de 20dB de la CO sur les 4000Hz (tous les audiogrammes sur anomalie de Mondini de son étude ayant cette particularité).
Les surdités post traumatiques
Les traumatismes de l’oreille responsables d’une surdité de transmission sont variés. Ces pathologies n’ont pas de caractère spécifique chez l’enfant.
Le blast auriculaire chez l’enfant fait principalement suite à des traumatismes à type de gifle ou à un baiser. Il en résulte des ruptures tympaniques et des atteintes ossiculaires. Le seuil de rupture tympanique se situe entre 50 et 160 kilopascals. Les tympans cicatriciels, plus fragiles, peuvent se rompre dès 40 kilopascals. La chaine ossiculaire peut subir des luxations (surtout incudostapédiennes, plus rarement stapédovestibulaires), des fractures (superstructure de l’étrier, branche descendante de l’enclume, col du marteau).
Les fractures du rocher peuvent survenir au cours d’accidents de la voie publique, de sports ou de chutes parfois apparemment bénignes. Elles peuvent passer inaperçues chez l’enfant et se révéler tardivement, par une surdité de transmission par luxation incudomalléaire ou incudostapédienne ou post traumatique par inclusion d’un fragment d’épiderme.
Les traumatismes directs tympano-ossiculaires sont fréquents : il s’agit du classique traumatisme par un coton-tige, ou par un crayon, responsable de perforation voire de traumatisme de la chaine ossiculaire. Les perforations sont, en général, de petite taille. En revanche, les atteintes ossiculaires peuvent être de tout type et le scanner des rochers peut les mettre en évidence avant l’exploration chirurgicale.
L’otospongiose juvénile
Cette pathologie est exceptionnelle, longtemps surévaluée aux dépends de l’aplasie mineure. L’aggravation de l’hypoacousie de transmission signe le diagnostic de l’otospongiose juvénile à la différence des aplasies mineures. Le scanner est indispensable, notamment pour éliminer une malformation de l’oreille interne avant platinotomie. Le traitement chirurgical est identique à celui des formes adultes, avec une réserve pour la pose d’un piston transplatinaire avant l’âge de 10 ans.
La maladie de Lobstein
Cette pathologie héréditaire autosomique dominante, responsable d’une fragilité osseuse et d’hyperlaxité ligamentaire, peut s’associer à une ankylose stapédo-vestibulaire. La platine apparait fine et fixée en périphérie avec, parfois, un étrier grêle et atrophié. Dans certains cas, il peut exister un épaississement platinaire avec un comblement osseux de la fenêtre ovale.
Source :
Les monographies du CCA Groupe – Surdité de l’enfant – 2003.
E.N Garabédian, F. denoyelle , R. Dauman , J.M Triglia , E. Tuy , N. Loudnon , P. Bouaziz , J. de Lorenzi








